
Introduction
When a Phase III trial closes, years of patient data, protocol decisions, deviations, and statistical analyses converge into a single document that regulators use to assess whether a new treatment meets the standard for approval. That document is the Clinical Study Report, commonly called the CSR. It is not a journal article, not an executive summary, and not a data appendix. It is the definitive, structured scientific account of everything that happened in a clinical trial, written to an international standard and submitted as part of the marketing authorization dossier.
Full Phase III CSRs frequently run into the hundreds of pages; in complex late-stage oncology or rare-disease trials, it is not unusual in industry practice for a report to exceed a thousand. That scale reflects what regulatory review requires: a complete, transparent, reproducible record from which agencies can independently evaluate the risk-benefit profile of an investigational product. Getting the document wrong, incomplete, or internally inconsistent does not just create rework. It can slow the review cycle, generate formal regulatory queries, or in serious cases, raise questions about trial integrity.
This guide covers what a CSR is, how it is structured under ICH E3, what types exist, who writes and reviews one, how it fits into the Common Technical Document (CTD), what regulatory changes are affecting it right now, and where AI-assisted drafting fits into the process.
Why the CSR Matters in Clinical Drug Development
The path from first-in-human dosing to a marketing application is long. The U.S. Department of Health and Human Services has cited mean Phase III durations of approximately 30.5 months and estimated that the full period from the start of clinical testing to marketing approval stretches over 90 months on average [1]. Phase III trials are also the most resource-intensive stage of development; published analyses estimating per-drug Phase III expenditure, when out-of-pocket and capital costs are combined, place those figures in the hundreds of millions [2].
"After that investment, the CSR is the document that converts trial conduct into regulatory evidence."
For applications that rely on clinical study evidence, CSRs are a core component of the clinical dossier. Per ICH M4E and its adoption by FDA, EMA, PMDA, and other ICH-aligned agencies, clinical study reports are placed in CTD Module 5, which contains the primary clinical evidence base for the dossier [3].
Beyond submission, CSRs serve several other functions across the product lifecycle. They are the primary contemporaneous scientific record of trial conduct and are referenced during GCP inspections and regulatory audits. CSR data also informs post-market surveillance studies and internal decisions about future development programs.
The ICH E3 Guideline: The Governing Standard
CSRs prepared for ICH-region regulatory submission are expected to follow ICH E3, "Structure and Content of Clinical Study Reports," originally approved in 1995 and effective since 1996 [4]. ICH E3 was developed to enable compilation of a single core CSR acceptable to all regulatory authorities across ICH member regions, meaning a document structured according to E3 can be submitted to the FDA, EMA, PMDA, and other ICH-aligned agencies simultaneously [5].
ICH E3 is guidance, not a rigid template. According to the ICH E3 Questions and Answers document (R1, 2012) [6], each report should consider all described topics unless clearly not relevant, but the specific sequence and grouping of topics may be adapted where an alternative structure is more logical for a particular study. Demographic baseline information, for example, may sit within the efficacy section or as a standalone section when the efficacy and safety populations differ substantially.
What is not flexible is completeness. Regulatory authorities expect a clear, complete, transparent, and reproducible account of the study. In its 2024 AI Observatory Report, the EMA noted that applicants are investigating generative AI tools to assist in drafting regulatory documentation, including the Clinical Study Report, itself a reflection of how resource-intensive CSR production has become [7]. The standard against which any such assistance is measured remains ICH E3.
ICH E3 was written primarily for interventional efficacy and safety trials of the type that were common in the mid-1990s; the 2012 Q&A supplemented it to address more complex modern designs [6]. In practice, many medical writing teams also work with CORE Reference (Clarity and Openness in Reporting: E3-based), a freely available user manual developed by EMWA and AMWA members to help authors navigate ICH E3 and regional guidance while producing CSRs that are as public-disclosure-ready as possible [8]. CORE Reference does not replace ICH E3; it provides content suggestions and best practices for applying E3 in today's more diverse study designs.
Structure of a Clinical Study Report
Under ICH E3, a full CSR follows a defined architecture covering every material aspect of trial conduct and results.
Title Page and Synopsis. The title page identifies the investigational product, protocol number, sponsor, principal or coordinating investigator, and report date. The synopsis provides a two-to-three-page summary of trial objectives, design, patient population, endpoints, and main efficacy and safety results. Because the synopsis must be intelligible as a standalone document within the CTD, it receives close early attention during regulatory review [6].
Table of Contents. A complete, navigable map of all sections, appendices, figures, and listings. In eCTD submissions this becomes part of the structured document architecture regulators navigate electronically.
Ethics and Administrative Structure. Documentation of ethics committee approvals, investigator names and affiliations, and descriptions of the sponsor, CROs, and contracted parties. These sections allow regulators to trace accountability for trial conduct.
Introduction and Study Objectives. The therapeutic context, scientific rationale, and clearly defined primary and secondary objectives. The endpoints tied to those objectives appear here.
Investigational Plan. Overall trial design (randomized, controlled, open-label, blinded), study population, inclusion and exclusion criteria, dosing regimen, statistical analysis plan (SAP), sample size justification, and planned monitoring approach.
Efficacy Evaluation. Data summaries, statistical results, and narrative conclusions for each efficacy endpoint. Tables, listings, and figures generated from the clinical and statistical analysis are included inline or referenced from appendices.
Safety Evaluation. Adverse event data, serious adverse events, laboratory findings, vital sign changes, and relevant subgroup analyses. The safety narrative must be coherent and traceable to the underlying datasets.
Appendices. Appendix 16 under ICH E3 contains the technical documentation underpinning the report: the protocol, protocol amendments, sample case report forms, ethics approvals, investigator information, and individual patient data listings where required. Some appendix content is region-specific and should be confirmed against local regulatory requirements before finalizing the document [6].
The practical consequence of this architecture is that a full Phase III CSR draws simultaneously on contributions from clinical operations, biostatistics, data management, pharmacovigilance, and medical writing. Coordinating those inputs without introducing internal inconsistencies is among the most persistent operational challenges in CSR production.
Types of Clinical Study Reports
ICH E3 and its supplementary Q&A acknowledge that reporting depth should suit the specific study, and regulatory medical writing practice, informed by CORE Reference and regional regulatory experience, recognizes several commonly used report formats [6][8][9].
Full CSR
Contains all sections and appendices described in ICH E3 and is the standard document for pivotal trials supporting a marketing authorization.
Abbreviated CSR
Used for studies not designed to demonstrate treatment effectiveness, such as pharmacokinetic studies, drug interaction studies, or Phase I single-dose safety studies.
Synoptic CSR
Covers trials that provide safety data without reporting on treatment effectiveness or clinical pharmacology.
Interim CSR
Documents trial progress at a pre-specified analysis point before the primary endpoint is reached.
Supplemental CSR
Adds detail to a completed full CSR. It may be required for planned-but-delayed analyses, unplanned exploratory analyses, or cross-study analyses not ready at the time of original submission.
A full CSR contains all sections and appendices described in ICH E3 and is the standard document for pivotal trials supporting a marketing authorization. It is the expected format for Phase III efficacy and safety trials placed in CTD Module 5, though the precise dossier requirements for any given application depend on the region, the study's role in the submission, and agency-specific expectations.
An abbreviated CSR is used for studies not designed to demonstrate treatment effectiveness, such as pharmacokinetic studies, drug interaction studies, or Phase I single-dose safety studies. These reports include complete datasets and safety tables but incorporate cross-references to the main CSR rather than standalone narrative sections [9].
A synoptic CSR covers trials that provide safety data without reporting on treatment effectiveness or clinical pharmacology. This compact format is appropriate for certain early-phase studies and post-approval pharmacovigilance studies [9].
An interim CSR documents trial progress at a pre-specified analysis point before the primary endpoint is reached. These arise in adaptive trial designs or when interim data are needed for a Data Safety Monitoring Board decision [9].
A supplemental CSR adds detail to a completed full CSR. It may be required for planned-but-delayed analyses, unplanned exploratory analyses, or cross-study analyses not ready at the time of original submission [8].
Identifying the correct format before writing begins determines the document shell, required appendices, and the level of narrative expected from the writing team.
What a Medical Writer Needs Before Starting a CSR
CSR production cannot begin in earnest without a defined set of source documents. Experienced authors recommend assembling these inputs before or immediately after database lock [10]:
Clinical & Regulatory Foundation
The final protocol and all amendments, the final SAP, the randomization and blinding documentation, the investigator list and ethics approvals, and any regulatory meeting minutes relevant to the trial design or endpoint strategy.
Statistical Outputs
The final, QC-checked tables, figures, and listings (TFLs) generated from the clean dataset, the final analysis datasets, and a clear population definitions document that maps each analysis set to the protocol criteria.
Safety & Administrative Docs
The coded, reconciled adverse event and serious adverse event listings, the protocol deviations log with an assessment of each deviation's impact, the final patient narratives for SAEs and discontinuations, sample case report forms or eCRF screenshots, the final informed consent form, and any Data Safety Monitoring Board correspondence or interim analysis reports relevant to the final CSR narrative.
The clinical and regulatory foundation covers the final protocol and all amendments, the final SAP, the randomization and blinding documentation, the investigator list and ethics approvals, and any regulatory meeting minutes relevant to the trial design or endpoint strategy.
The statistical outputs include the final, QC-checked tables, figures, and listings (TFLs) generated from the clean dataset, the final analysis datasets, and a clear population definitions document that maps each analysis set to the protocol criteria.
The safety and deviations record encompasses the coded, reconciled adverse event and serious adverse event listings, the protocol deviations log with an assessment of each deviation's impact, and the final patient narratives for SAEs and discontinuations.
The administrative documentation includes sample case report forms or eCRF screenshots, the final informed consent form, and any Data Safety Monitoring Board correspondence or interim analysis reports relevant to the final CSR narrative.
Gaps in any of these categories after database lock are among the most common causes of extended CSR timelines. A writing team that waits months for a finalized SAP or clean TFLs will take months longer to deliver a submission-ready document.
Who Writes and Reviews a CSR?
A CSR involves a larger cross-functional team than many sponsors anticipate. The primary author is typically a senior regulatory medical writer, in-house or contracted through a CRO or specialist medical writing firm. The writer's role goes beyond transcribing data. They translate statistical outputs and clinical observations into a coherent regulatory narrative that accurately represents trial conduct and conclusions.
The responsible biostatistician confirms that tables, listings, and figures align with the pre-specified SAP and signs off on statistical sections. The principal or coordinating investigator may also be required to sign the completed CSR depending on submission region; their signature page goes in Appendix 16.1.5 under ICH E3 [4]. Clinical pharmacology, safety, regulatory affairs, and data management teams typically participate in the review cycle. A robust CSR undergoes multiple rounds of medical review, statistical review, and editorial quality control before the approved version is submitted to a regulatory authority.
Starting CSR preparation early is consistently recommended by experienced medical writers. Initiating the document shell before last patient last visit (LPLV), and ensuring the finalized SAP, clean datasets, protocol deviations log, and AE coding are delivered to the writing team promptly after database lock, shortens overall writing timelines and reduces the risk of narrative-to-data inconsistencies that arise when reporting begins months after a trial has closed [10].
Common CSR Quality Control Checks
Before a CSR goes to a regulatory authority, the document should pass through a structured QC cycle that checks the following areas:
Protocol and SAP alignment
Every efficacy and safety endpoint described in the results sections must map to an objective stated in the protocol and a corresponding analysis pre-specified in the SAP. Any post-hoc or unplanned analysis must be clearly identified as such.
Population definitions
The full analysis set, per-protocol set, and safety population must be defined consistently across the methods, statistical sections, TFLs, and narrative. Inconsistencies in population labeling are a frequent source of regulatory questions.
TFL traceability
Table, figure, and listing numbers cited in the text must match those in the appendices. Row totals, subject counts, and summary statistics must reconcile across tables that draw from the same dataset.
Safety narrative completeness
Every serious adverse event and every discontinuation due to an adverse event should have a patient narrative. AE tables should reconcile with the SAE listings, and any discrepancies should be resolved and documented before submission.
Protocol deviation handling
The deviations log should be reflected accurately in the methods section, and the impact assessment for major deviations should appear in the efficacy and safety narratives where relevant.
Appendix completeness
Appendix 16 items required for the submission region (protocol, amendments, ethics approvals, investigator list, sample CRFs) should be confirmed against regional guidance before the document is locked.
Module 2.7 consistency
Key efficacy and safety conclusions in the CSR should be cross-checked against the summary of clinical efficacy (Section 2.7.3) and summary of clinical safety (Section 2.7.4). Numerical and narrative mismatches between Module 5 and Module 2 are a recognized source of regulatory review friction [3].
Regulatory and Documentation Considerations
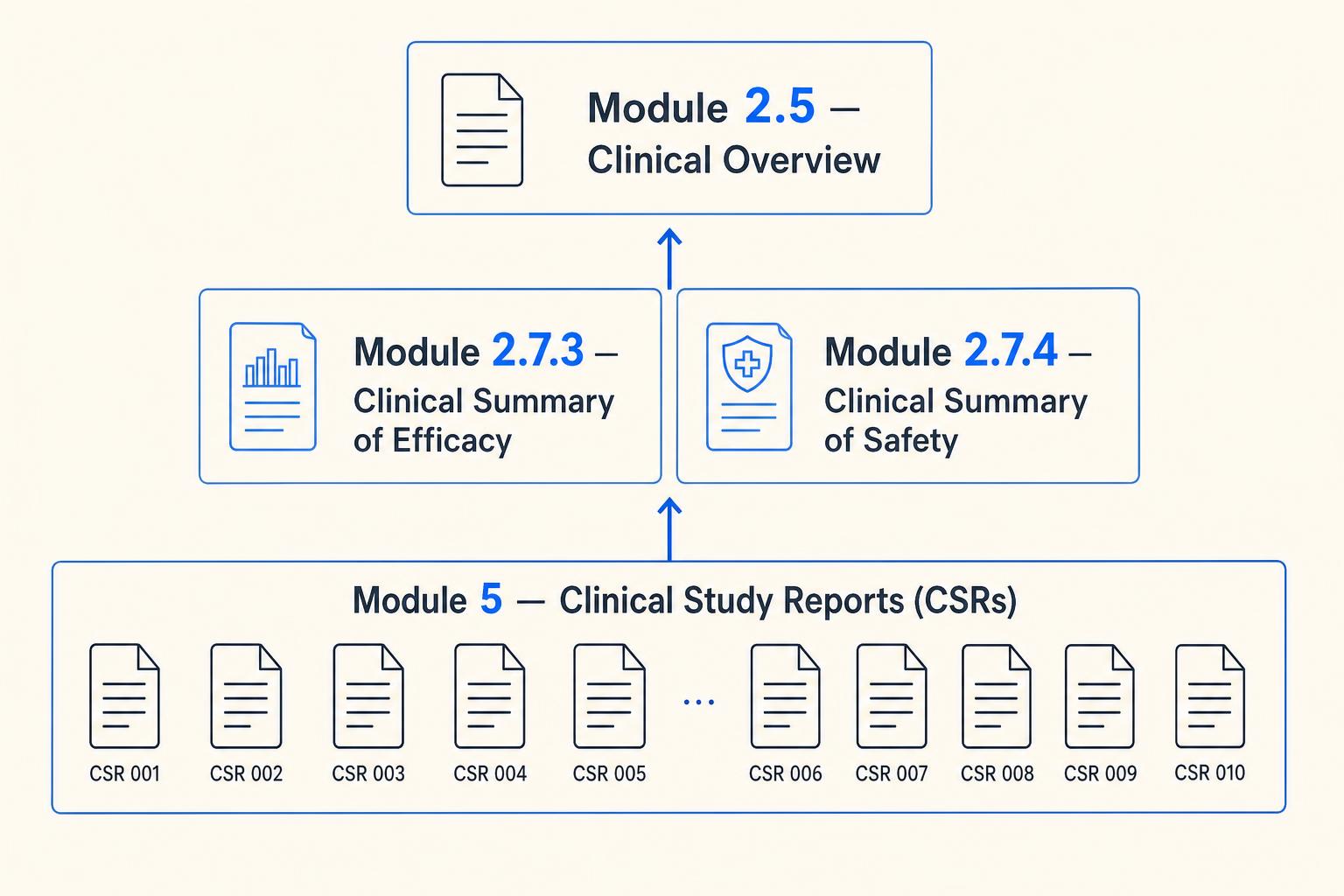
Placement in the CTD
Under the Common Technical Document format, clinical study reports are placed in Module 5, organized according to ICH M4E [3]. The individual CSRs provide the primary clinical evidence base from which the clinical overview (Module 2.5) and clinical summaries (Module 2.7) are synthesized. Specifically, the summary of clinical efficacy (Section 2.7.3) and summary of clinical safety (Section 2.7.4) draw directly from CSR findings. Inconsistencies between CSR data and Module 2.7 summaries are a recognized source of review friction, and discrepancies between what appears in Module 2 and the underlying Module 5 reports prompt questions from reviewers.
AI and Automation in CSR Production
CSR production is time-intensive, requires sustained coordination across large cross-functional teams, and is vulnerable to the cross-document inconsistencies that arise when different groups work from shared datasets on overlapping timelines. AI tools are being applied to specific, bounded tasks within this workflow.
Patient narrative generation, population of standard table shells, and production of listings from structured clinical data are the most active areas. In its 2024 AI Observatory Report, the EMA confirmed that applicants are investigating generative AI to assist in drafting regulatory documentation including the CSR [7]. The report frames human oversight, validation, and quality review as continuing regulatory expectations regardless of the tools used.
What AI cannot currently replace is the medical and regulatory judgment required to characterize safety signals accurately, interpret efficacy findings, and ensure the CSR narrative coheres into a defensible regulatory position consistent with the protocol, SAP, and the broader CTD. As of mid-2026, I could not identify dedicated FDA or EMA guidance specific to AI-generated CSR content, though existing regulatory accountability and validation expectations apply to any tool used in submission document production. Sponsors using AI drafting tools should verify the tool has been validated for the specific study type and therapeutic area, ensure all outputs are reviewed by a qualified medical writer against source data and the SAP before inclusion, and maintain a clear document provenance audit trail. Sponsors should be prepared to explain how any regulatory document was produced if the question arises during a GCP inspection.
How Kitsa Fits Into This Problem
Generating a submission-ready CSR requires more than a good template. It requires access to the right data at the right time, alignment with the protocol and SAP, cross-document consistency with the broader CTD, and a structured writing process that can move from database lock to a final approved document without losing clinical context. Kitsa's KScribe platform is built to support exactly this workflow: AI-native regulatory document generation that produces ICH E3-structured CSR drafts from trial data, maintains consistency with the protocol and other submission documents, and reduces the manual coordination burden on writing teams and regulatory affairs. For sponsors and CROs managing multiple submissions simultaneously, structured automation at the document layer is where meaningful time savings are achievable without compromising the human oversight that regulatory agencies require.
Key Takeaways
- •A Clinical Study Report (CSR) is a comprehensive regulatory document describing a clinical trial's methodology, conduct, and results, structured according to ICH E3. It is a core component of the clinical dossier for NDA, BLA, and MAA applications.
- •ICH E3 governs CSR structure globally, enabling a single document to be submitted to FDA, EMA, and other ICH-aligned agencies. The guideline offers flexibility in organization but expects all relevant topics to be addressed. CORE Reference provides a complementary practical user manual for applying E3 in complex modern study designs.
- •Full, abbreviated, synoptic, interim, and supplemental are commonly used CSR formats in regulatory medical writing practice, reflecting different study purposes and reporting needs. Identifying the correct format before writing begins determines the document shell, required appendices, and narrative depth.
- •CSRs are placed in CTD Module 5 per ICH M4E. Their content feeds the clinical overview (Module 2.5) and clinical summaries (Module 2.7). Consistency across these documents is a baseline regulatory expectation.
- •For FDA NDA submissions, ISS and ISE are required documents under 21 CFR 314.50, placed in Module 5, Section 5.3.5.3. EMA submissions handle cross-trial integration through Module 2.7. Both follow the CTD structure but differ on these specific requirements.
- •FDA began supporting eCTD v4.0 for new NDA, BLA, ANDA, IND, and MF submissions from September 16, 2024. Both v3.2.2 and v4.0 remain accepted as of mid-2026; mandatory v4.0-only adoption has not been scheduled.
- •Under EU-CTR Article 37(4), the CSR for any trial used in an EU marketing authorization application must be submitted to CTIS within 30 days of the marketing authorization decision. Summary results and lay summaries carry earlier, separate deadlines.
KScribe · AI Regulatory Document Generation
From database lock to final approved CSR; with structure intact.
Generate ICH E3-structured CSR drafts from trial data; with protocol alignment, cross-document consistency, and a structured writing process from database lock to final approved document.
Frequently Asked Questions
References
- [1] U.S. Department of Health and Human Services, Office of the Assistant Secretary for Planning and Evaluation. "Examination of Clinical Trial Costs and Barriers for Drug Development." ASPE, 2014. https://aspe.hhs.gov/reports/examination-clinical-trial-costs-barriers-drug-development-0
- [2] DiMasi JA, Grabowski HG, Hansen RW. "Innovation in the pharmaceutical industry: New estimates of R&D costs." Journal of Health Economics. 2016;47:20-33. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4951205/
- [3] ICH. "M4E(R2): The Common Technical Document for the Registration of Pharmaceuticals for Human Use: Efficacy." ICH, 2016. https://www.fda.gov/media/93569/download
- [4] ICH. "E3: Structure and Content of Clinical Study Reports." International Council for Harmonisation, 1995 (effective 1996). https://database.ich.org/sites/default/files/E3_Guideline.pdf
- [5] European Medicines Agency. "ICH E3 Structure and Content of Clinical Study Reports: Scientific Guideline." EMA. https://www.ema.europa.eu/en/ich-e3-structure-content-clinical-study-reports-scientific-guideline
- [6] ICH. "E3: Structure and Content of Clinical Study Reports, Questions and Answers (R1)." ICH, 2012. https://database.ich.org/sites/default/files/E3_Q&As_R1_Q&As.pdf
- [7] European Medicines Agency. "2024 AI Observatory Report." EMA, May 2025. https://www.ema.europa.eu/en/documents/report/2024-ai-observatory-report_en.pdf
- [8] Hamilton S, Bernstein AB, Blakey G, et al. "Developing the Clarity and Openness in Reporting: E3-based (CORE) Reference user manual for creation of clinical study reports in the era of clinical trial transparency." Research Integrity and Peer Review. 2016;1:4. https://pmc.ncbi.nlm.nih.gov/articles/PMC5794039/
- [9] Taranum S. "Clinical study reports: A snapshot for aspiring medical writers." Medical Writing. 2023;32(1):70-74. https://doi.org/10.56012/qett4705
- [10] Bhatt C, Seger D. "Effective authoring of clinical study reports: A companion guide." Medical Writing. 2014;23(3):162-167. https://journal.emwa.org/regulatory-writing-basics/effective-authoring-of-clinical-study-reports-a-companion-guide/
- [11] FDA. "Guidance for Industry: Integrated Summaries of Effectiveness and Safety -- Location Within the Common Technical Document." U.S. Food and Drug Administration. https://www.fda.gov/files/drugs/published/Integrated-Summaries-of-Effectiveness-and-Safety--Location-Within-the-Common-Technical-Document.pdf
- [12] FDA. "Electronic Common Technical Document (eCTD)." U.S. Food and Drug Administration; Federal Register Notice, September 16, 2024. https://www.fda.gov/drugs/electronic-regulatory-submission-and-review/electronic-common-technical-document-ectd
- [13] European Commission. "Clinical Trials -- Regulation (EU) No 536/2014." European Commission Health, Medicinal Products. https://health.ec.europa.eu/medicinal-products/clinical-trials/clinical-trials-regulation-eu-no-5362014_en
- [14] European Medicines Agency. "CTIS Public Portal: Trial Results." EMA, December 2024. https://www.ema.europa.eu/en/documents/other/clinical-trial-information-system-ctis-public-portal-trial-results_en.pdf
- [15] European Union. "Regulation (EU) No 536/2014 of the European Parliament and of the Council, Article 37(4)." EUR-Lex, 2014. https://eur-lex.europa.eu/legal-content/EN/TXT/HTML/?uri=CELEX:32014R0536
- [16] European Medicines Agency. "Revised CTIS Transparency Rules." EMA, October 2023. https://www.ema.europa.eu/en/documents/other/revised-ctis-transparency-rules_en.pdf